

license: other
AIDO.RAGProtein-16B
AIDO.RAGProtein-16B is a multimodal protein language model that integrates MSA and structural data based on the AIDO.Protein-16B model. Its training is divided into multiple stages, 100 billion tokens are trained on UniRef50/UniClust30 MSA data, and 80 billion tokens are trained on AlphaFold Database MSA and structural data.
Model Architecture Details
AIDO.RAGProtein-16B is a transformer encoder-only architecture with the dense MLP layer in each transformer block replaced by a sparse MoE layer. It uses single amino acid tokenization and is optimized using a masked languange modeling (MLM) training objective. For each token, 2 experts will be selectively activated by the top-2 rounting mechiansim.
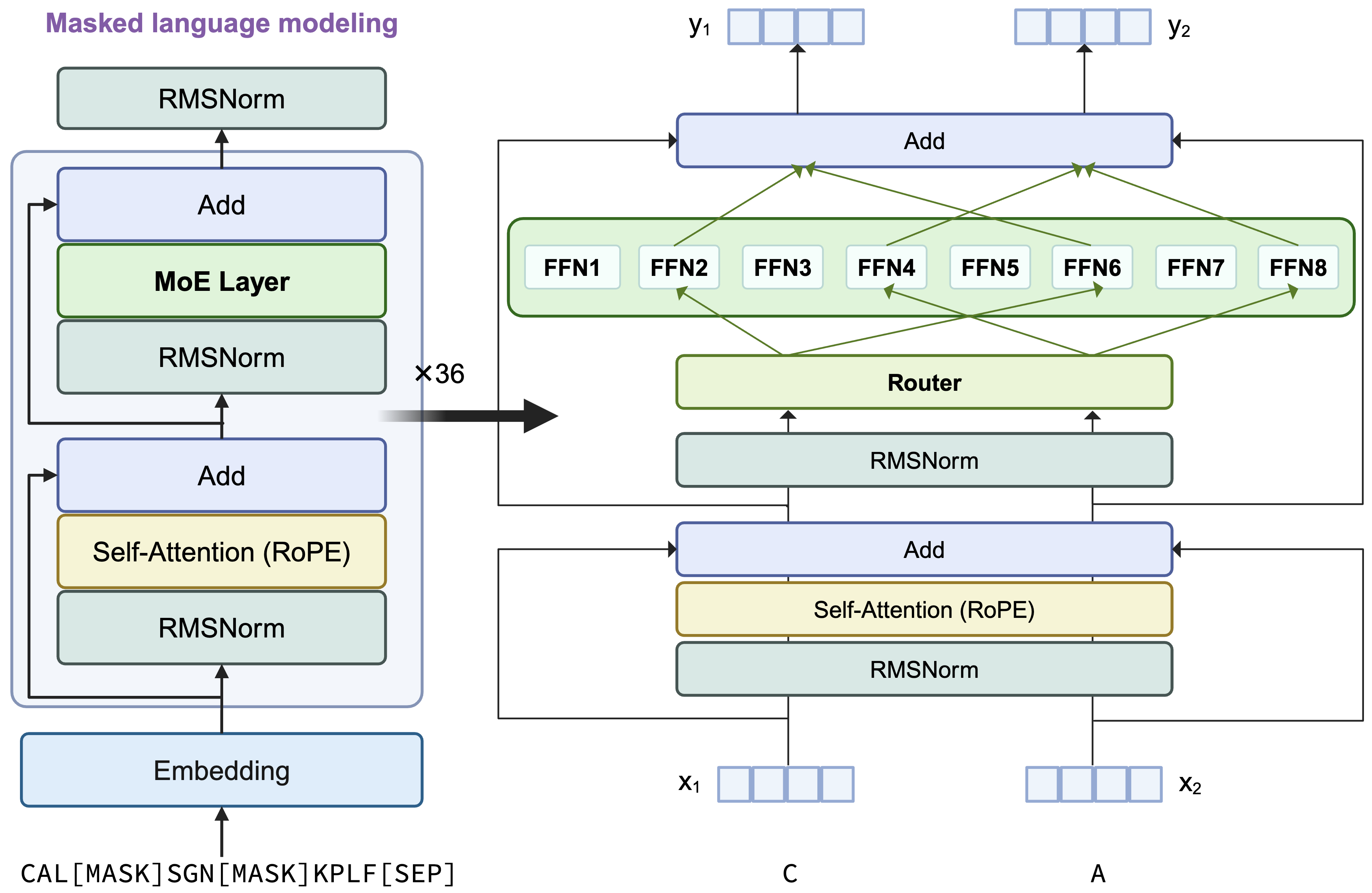
More architecture details are shown below:
| Model Arch Component | Value |
|---|---|
| Num Attention Head | 36 |
| Num Hidden Layer | 36 |
| Hidden Size | 2304 |
| FFN Hidden Size | 7680 |
| Num MoE Layer per Block | 8 |
| Num MoE Layer per Token | 2 |
| Vocab Size | 44 |
| Context Length | 2048 |
Pre-training of AIDO.RAGProtein-16B
Here we briefly introduce the details of pre-training of AIDO.RAGProtein-16B. Mainly divided into three stages: (1) 1D -> 2D finetuning; (2) UniRef50/Uniclust30 MSA finetuning; (3) AlphaFold Database MSA & Structure tokens finetuning.
Data
UniRef50/Uniclust30 MSA dataset: We utilized sequences from UniRef50 as queries to search for homologous sequences in UniClust30, subsequently constructing multiple sequence alignments (MSAs). UniRef50 comprises a total of 53.6 million sequences. Using HHblits, we searched all sequences, identifying over 25 homologous sequences for 23.7 million of them. This dataset was directly used as the training set, referred to as HHblits_MSA. The remaining 29.9 million sequences were input into MSA Retriever, resulting in 7.7 million sequences with more than 25 homologous sequences. This dataset was designated as Retriever_MSA. During training, RAGPLM randomly sampled from the two datasets with probabilities of 0.75 and 0.25
AlphaFold Database MSA & Structure dataset: We downloaded all the structural data from the AlphaFold Database and only kept the structures where the amino acid ratio of pLDDT>70 was greater than 40%. Then we used mmseqs to cluster the remaining sequences with seq id=0.5, and retained a representative sequence for each class. Final we get 46.9 million sequence/structure pairs. For each structure, we used genbio-ai/AIDO.StructureTokenizer to obtain the corresponding structure tokens and structure embedding. And used MSA Retriever to obtain the MSA corresponding to the sequence.
Training Details
Model training is divided into three stages:
(1) 1D -> 2D finetuning:
Same training data with AIDO.Protein-16B, but use 2D rotary position embedding to encode the tokens;
(2) UniRef50/Uniclust30 MSA finetuning
We used UniRef50/Uniclust30 MSA dataset to finetune the model from stage (1). Refer AIDO.RAGPLM for more information.
(3) AFDB MSA & Structure tokens finetuning:
We fine-tuned a pretrained masked language model using MSA data by concatenating the query sequence with homologous sequences. The input structure embedding (hidden dimension 384) is linearly mapped to 2304 and then added to the corresponding embedding of the query sequence tokens.
Mask of sequences: We introduced several modifications to the standard BERT masking strategy: (1) We randomly sampled 0.05×L span positions from a query sequence of length L, with span lengths following a geometric distribution (p=0.2), and capped the maximum length at 10. Our experiments revealed that this settings lead to an average of 15% of the query tokens were masked. (2) To prevent information leakage, when a residue was selected, all residues at the same index across all sequences (the column of the MSA matrix) were also masked. (3) When a column of MSA was selected for masking, the entire column was replaced with the <MASK> token in 80% of cases, with random amino acids in 10% of cases, and remained unchanged in the remaining 10% of cases.
Mask of structure: In 20% of the cases, we randomly replaced the structure embedding with 0; in 80% of the cases, we randomly sampled a certain number of amino acids using the BetaLinear30 distribution and masked their structure embedding. The BetaLinear30 distribution is defined as a combination of 20% of the [0, 1] uniform distribution and 80% of the Beta(3, 9) Beta distribution.
Positional embedding: To help the model distinguish which tokens are from the same chain and which tokens have the same residue index, we use 2D rotary position embedding to encode the tokens.
Loss: The loss function consists of a sequence loss function and a structure loss function (weights are 1.0 and 0.01 respectively). The sequence loss function is the CrossEntropy function that recovers the masked sequence tokens, and the structure loss function is the CrossEntropy function that predicts each masked structure token.
| Hyper-params | (1) 1D -> 2D finetuning | (2) UniRef50/Uniclust30 MSA finetuning | (3) AFDB MSA & Structure tokens finetuning |
|---|---|---|---|
| Data | ColabFoldDB, UniRef | HHblits_MSA, Retriever_MSA | AFDB MSA & Structure tokens |
| Global Batch Size | 512 | 256 | 256 |
| Sequence length | 2048 | 12800 | 12800 |
| Per Device Micro Batch Size | 1 | 1 | 1 |
| Precision | Mixed FP32-FP16 | Mixed FP32-FP16 | Mixed FP32-FP16 |
| LR | [5e-6,5e-5] | [1e-6, 1e-5] | 1e-5 |
| Num Tokens | 10 billion | 100 billion | 80 billion |
Tokenization
We encode protein sequence with single amino acid resolution with 44 vocabularies, where 24 tokens represent amino acid types and 20 are special tokens. Sequences were also suffixed with a [SEP] token as hooks for downstream tasks.
How to Use
Build any downstream models from this backbone with ModelGenerator
For more information, visit: Model Generator
mgen fit --model SequenceClassification --model.backbone aido_ragprotein_16b --data SequenceClassificationDataModule --data.path <hf_or_local_path_to_your_dataset>
mgen test --model SequenceClassification --model.backbone aido_ragprotein_16b --data SequenceClassificationDataModule --data.path <hf_or_local_path_to_your_dataset>
Or use directly in Python
Embedding
import torch
from modelgenerator.tasks import Embed
model = Embed.from_config({"model.backbone": "aido_protein_16b_ragplm"}).eval()
model.backbone.max_length = 12800
data = torch.load("ModelGenerator/experiments/AIDO.RAGPLM/examples.pt", 'cpu')[0]
transformed_batch = model.transform(data)
with torch.no_grad():
embedding = model(transformed_batch)
print(embedding.shape)
Sequence Level Classification
import torch
from modelgenerator.tasks import SequenceClassification
model = SequenceClassification.from_config({"model.backbone": "aido_protein_16b_ragplm", "model.n_classes": 2}).eval()
model.backbone.max_length = 12800
data = torch.load("ModelGenerator/experiments/AIDO.RAGPLM/examples.pt", 'cpu')[0]
transformed_batch = model.transform(data)
with torch.no_grad():
logits = model(transformed_batch)
print(logits)
print(torch.argmax(logits, dim=-1))
Token Level Classification
import torch
from modelgenerator.tasks import TokenClassification
model = TokenClassification.from_config({"model.backbone": "aido_protein_16b_ragplm", "model.n_classes": 3}).eval()
model.backbone.max_length = 12800
data = torch.load("ModelGenerator/experiments/AIDO.RAGPLM/examples.pt", 'cpu')[0]
transformed_batch = model.transform(data)
with torch.no_grad():
logits = model(transformed_batch)
print(logits)
print(torch.argmax(logits, dim=-1))
Regression
from modelgenerator.tasks import SequenceRegression
model = SequenceRegression.from_config({"model.backbone": "aido_protein_16b_ragplm"}).eval()
model.backbone.max_length = 12800
data = torch.load("ModelGenerator/experiments/AIDO.RAGPLM/examples.pt", 'cpu')[0]
transformed_batch = model.transform(data)
with torch.no_grad():
logits = model(transformed_batch)
print(logits.shape)
Citation
Please cite AIDO.RAGProtein-16B using the following BibTex code:
@inproceedings{sun_mixture_2024,
title = {Mixture of Experts Enable Efficient and Effective Protein Understanding and Design},
url = {https://www.biorxiv.org/content/10.1101/2024.11.29.625425v1},
doi = {10.1101/2024.11.29.625425},
publisher = {bioRxiv},
author = {Sun, Ning and Zou, Shuxian and Tao, Tianhua and Mahbub, Sazan and Li, Dian and Zhuang, Yonghao and Wang, Hongyi and Cheng, Xingyi and Song, Le and Xing, Eric P.},
year = {2024},
booktitle={NeurIPS 2024 Workshop on AI for New Drug Modalities},
}
@article {Li2024.12.02.626519,
author = {Li, Pan and Cheng, Xingyi and Song, Le and Xing, Eric},
title = {Retrieval Augmented Protein Language Models for Protein Structure Prediction},
url = {https://www.biorxiv.org/content/10.1101/2024.12.02.626519v1},
year = {2024},
doi = {10.1101/2024.12.02.626519},
publisher = {bioRxiv},
booktitle={NeurIPS 2024 Workshop on Machine Learning in Structural Biology},
}