
AIDO.Protein-RAG-16B-proteingym-dms-zeroshot
AIDO.Protein-RAG-16B-proteingym-dms-zeroshot is a multimodal protein language model that integrates Multiple Sequence Alignment (MSA) and structural data, building upon the AIDO.Protein-16B foundation. The training process comprises three main stages:
- 2D RoPE encoding fine-tuning
- Initial training on 100 billion tokens from UniRef50/UniClust30 MSA data
- Subsequent training on 23 billion tokens from AlphaFold Database MSA and structural data
Model Architecture
AIDO.Protein-RAG-16B-proteingym-dms-zeroshot employs a transformer encoder-only architecture featuring sparse Mixture-of-Experts (MoE) layers that replace dense MLP layers in each transformer block. Utilizing single amino acid tokenization and optimized through masked language modeling (MLM), the model activates 2 experts per token via top-2 routing mechanisms.
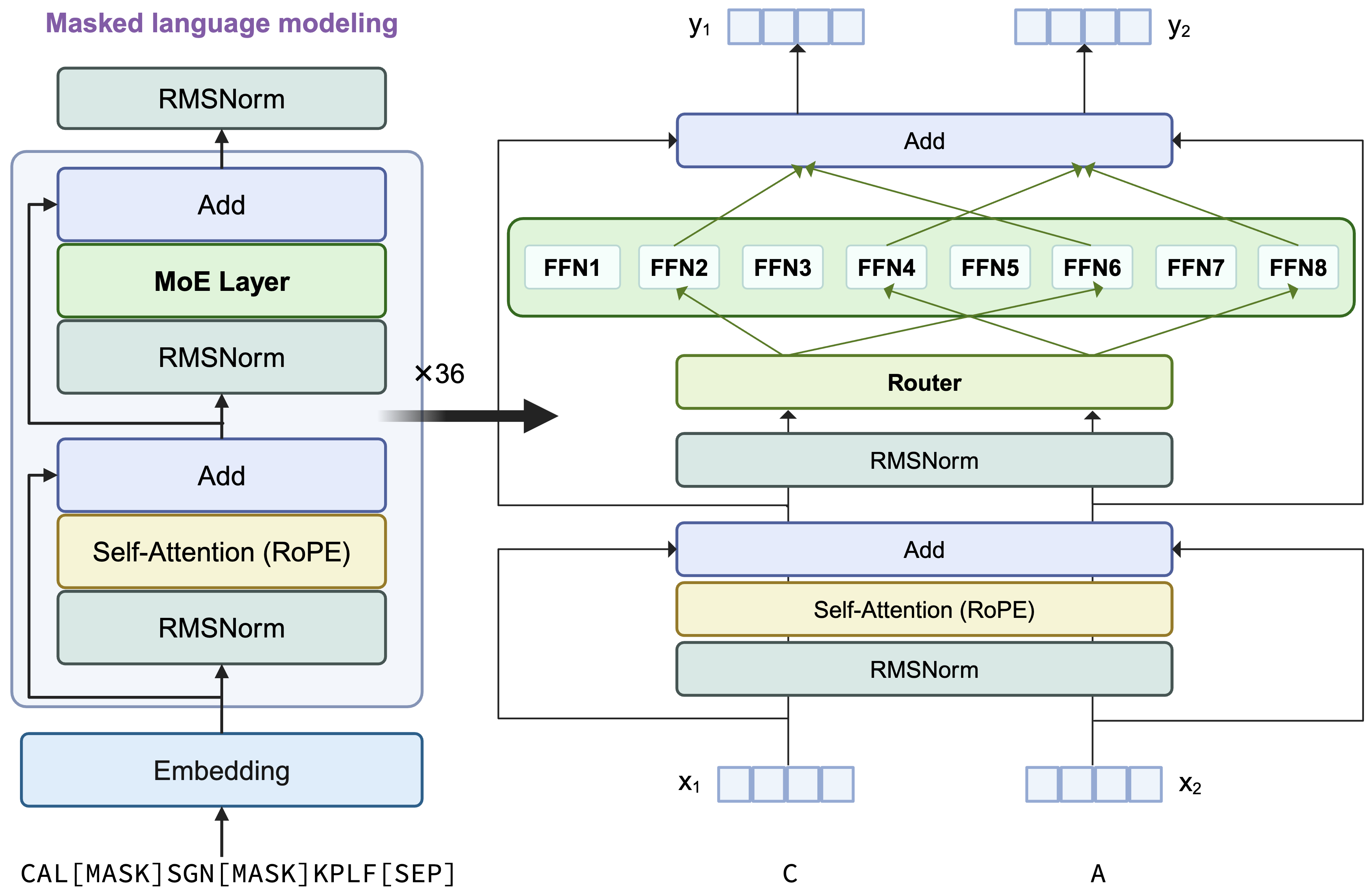
More architecture details are shown below:
| Model Arch Component | Value |
|---|---|
| Num Attention Head | 36 |
| Num Hidden Layer | 36 |
| Hidden Size | 2304 |
| FFN Hidden Size | 7680 |
| Num MoE Layer per Block | 8 |
| Num MoE Layer per Token | 2 |
| Vocab Size | 44 |
| Context Length | 2048 |
Pre-training of AIDO.Protein-RAG-16B-proteingym-dms-zeroshot
Here we briefly introduce the details of pre-training of AIDO.Protein-RAG-16B-proteingym-dms-zeroshot. Mainly divided into three stages: (1) 1D -> 2D RoPE encoding fine-tuning; (2) UniRef50/Uniclust30 MSA fine-tuning; (3) AlphaFold Database MSA & Structure tokens fine-tuning
Data
UniRef50/Uniclust30 MSA dataset: We utilized sequences from UniRef50 as queries to search for homologous sequences in UniClust30, subsequently constructing multiple sequence alignments (MSAs). UniRef50 comprises a total of 53.6 million sequences. Using HHblits, we searched all sequences, identifying over 25 homologous sequences for 23.7 million of them. This dataset was directly used as the training set, referred to as HHblits_MSA. The remaining 29.9 million sequences were input into MSA Retriever, resulting in 7.7 million sequences with more than 25 homologous sequences. This dataset was designated as Retriever_MSA. During training, RAGPLM randomly sampled from the two datasets with probabilities of 0.75 and 0.25. Refer to AIDO.Protein-RAG-3B paper (link) for more information.
AlphaFold Database MSA & Structure dataset: We downloaded all structural data from the AlphaFold Database and kept only those where more than 40% of amino acids had a pLDDT score > 70. The remaining sequences were clustered using mmseqs (seq id=0.5), and one representative per cluster was retained, resulting in 46.9 million sequence/structure pairs. For each structure, we used genbio-ai/AIDO.StructureTokenizer to obtain structure tokens and embeddings. MSA Retriever was used to obtain the corresponding MSA.
Training Details
Model training is divided into three stages:
(1) 1D -> 2D RoPE Encoding Fine-tuning
Same training data as AIDO.Protein-16B, but with 2D rotary position embedding for token encoding.
(2) UniRef50/UniClust30 MSA Fine-tuning
The model from Stage 1 is further fine-tuned on the UniRef50/Uniclust30 MSA dataset. See the AIDO.Protein-RAG-3B paper for more.
(3) AlphaFold Database MSA & Structure Fine-tuning
We fine-tuned the model with concatenated query and homologous sequences. Structure embeddings (dim = 384) are linearly mapped to 2304 and added to the query token embeddings.
Sequence Masking
Randomly sample
0.05 × Lspan positions from a query of lengthL. Span lengths follow a geometric distribution (p=0.2), capped at length 10. On average, ~15% of query tokens are masked.When a residue is selected, its aligned residues across all sequences (MSA column) are also masked.
For masked MSA columns: 80% are replaced with
<MASK>, 10% with random amino acids, and 10% left unchanged.
Structure Masking
In 20% of cases, structure embeddings are replaced with 0.
In 80% of cases, a number of amino acids is sampled using the BetaLinear30 distribution and corresponding embeddings are zeroed. (BetaLinear30 = 20% Uniform(0,1) + 80% Beta(3,9)).
Positional Embedding
We use 2D rotary position embedding to help the model distinguish token chain identities and residue indices. See AIDO.Protein-RAG-3B paper (link) for more information.
Loss Function
Total loss is a weighted sum of sequence loss (weight 1.0) and structure loss (weight 0.025).
Sequence loss: CrossEntropy loss for masked token prediction.
Structure loss: CrossEntropy loss for masked structure token prediction.
| Hyper-params | (1) 1D -> 2D fine-tuning | (2) UniRef50/Uniclust30 MSA fine-tuning | (3) AFDB MSA & Structure tokens fine-tuning |
|---|---|---|---|
| Initialized parameters | AIDO.Protein-16B | Stage (1) | Stage (2) |
| Data | ColabFoldDB, UniRef | HHblits_MSA, Retriever_MSA | AFDB MSA & Structure tokens |
| Global Batch Size | 512 | 256 | 256 |
| Sequence length | 2048 | 12800 | 12800 |
| Per Device Micro Batch Size | 1 | 1 | 1 |
| Precision | Mixed FP32-FP16 | Mixed FP32-FP16 | Mixed FP32-FP16 |
| LR | [5e-6,5e-5] | [1e-6, 1e-5] | 1e-5 |
| Num Tokens | 10 billion | 100 billion | 23 billion |
| Structural loss | N/A | N/A | 0.025 |
Tokenization
We encode protein sequence with single amino acid resolution with 44 vocabularies, where 24 tokens represent amino acid types and 20 are special tokens. Sequences were also suffixed with a [SEP] token as hooks for downstream tasks.
Results
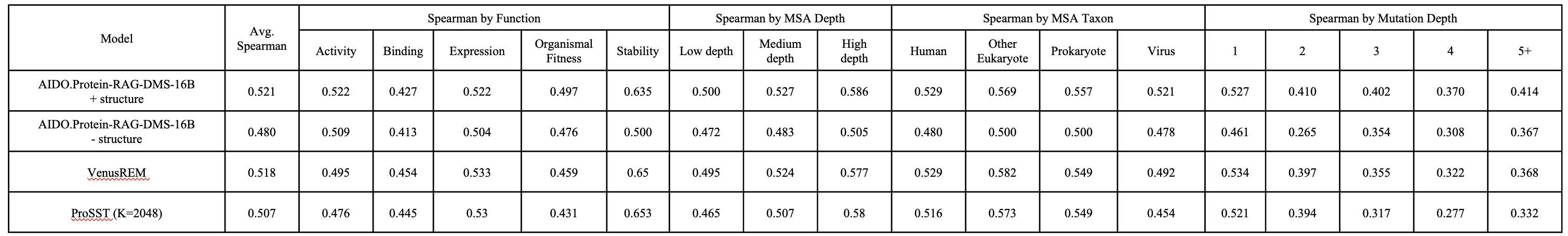
Zero-shot DMS score
How to Run
Load the model and tokenizer
from transformers import AutoModelForCausalLM, AutoTokenizer, AutoConfig, AutoModelForMaskedLM
tokenizer = AutoTokenizer.from_pretrained("genbio-ai/AIDO.Protein-RAG-16B-proteingym-dms-zeroshot", trust_remote_code=True)
model = AutoModelForCausalLM.from_pretrained("genbio-ai/AIDO.Protein-RAG-16B-proteingym-dms-zeroshot", trust_remote_code=True, torch_dtype=torch.bfloat16)
model = model.bfloat16().eval().to('cuda:0')
Clone the github respository and install environment TODO
Please read introduction of github respository to get the detail of installing environment and running method.
conda create -n ragplm python=3.11 -y
conda activate ragplm
pip install tabulate seaborn deepspeed
pip install git+https://github.com/genbio-ai/ModelGenerator.git
git clone https://github.com/genbio-ai/AIDO.Protein-RAG-proteingym-dms-zeroshot.git
cd AIDO.Protein-RAG-proteingym-dms-zeroshot
git clone https://huggingface.co/datasets/genbio-ai/ProteinGYM-DMS-RAG-zeroshot
mv ProteinGYM-DMS-RAG-zeroshot/msa_data ProteinGYM-DMS-RAG-zeroshot/*.gz .
tar xf dms_data.tar.gz
tar xf struc_data.tar.gz
mkdir output
Run zero-shot
python compute_fitness.py --dms_ids PTEN_HUMAN_Mighell_2018
Citation
Please cite AIDO.Protein-RAG-16B-proteingym-dms-zeroshot using the following BibTex code:
@inproceedings{sun_mixture_2024,
title = {Mixture of Experts Enable Efficient and Effective Protein Understanding and Design},
url = {https://www.biorxiv.org/content/10.1101/2024.11.29.625425v1},
doi = {10.1101/2024.11.29.625425},
publisher = {bioRxiv},
author = {Sun, Ning and Zou, Shuxian and Tao, Tianhua and Mahbub, Sazan and Li, Dian and Zhuang, Yonghao and Wang, Hongyi and Cheng, Xingyi and Song, Le and Xing, Eric P.},
year = {2024},
booktitle={NeurIPS 2024 Workshop on AI for New Drug Modalities},
}
@article {Li2024.12.02.626519,
author = {Li, Pan and Cheng, Xingyi and Song, Le and Xing, Eric},
title = {Retrieval Augmented Protein Language Models for Protein Structure Prediction},
url = {https://www.biorxiv.org/content/10.1101/2024.12.02.626519v1},
year = {2024},
doi = {10.1101/2024.12.02.626519},
publisher = {bioRxiv},
booktitle={NeurIPS 2024 Workshop on Machine Learning in Structural Biology},
}
- Downloads last month
- 211